Representations
When implementing an EA, one of the first design decisions that a practitioner must make is how to represent their problem in an individual. In this section we share how to structure individuals to represent a posed solution instance for a given problem.
Generally, each representation has a specific function, or set of functions, to create genomes for values of that representation type. There is sometimes also a decoders tailored to translate genomes into desired values. And, lastly, there will be a set of pipeline operators specific to that representation.
In summary, representations can have the following:
- initializers
These are for creating random genomes of that representation.
- decoders
These are for translating genomes to usable values; note not all representations need decoders in that you can directly use the genome values, and which is typical for real-valued and integer representations. (Which are a type of _phenotypic_ representation.)
- pipeline operators
Most all representations will have pipeline operators that are specific to that type
Binary representations
A common representation for individuals is as a string of binary digits.
Decoders for binary representations.
- class leap_ec.binary_rep.decoders.BinaryToIntDecoder(*descriptors)
Bases:
DecoderA decoder that converts a Boolean-vector genome into an integer-vector phenome.
- decode(genome, *args, **kwargs)
Converts a Boolean genome to an integer-vector phenome by interpreting each segment of the genome as low-endian binary number.
- Parameters
genome – a list of 0s and 1s representing a Boolean genome
- Returns
a corresponding list of ints representing the integer-vector phenome
For example, a Boolean representation of [1, 12, 5] can be decoded like this:
>>> import numpy as np >>> d = BinaryToIntDecoder(4, 4, 4) >>> b = np.array([0,0,0,1, 1, 1, 0, 0, 0, 1, 1, 0]) >>> d.decode(b) array([ 1, 12, 6])
- class leap_ec.binary_rep.decoders.BinaryToIntGreyDecoder(*descriptors)
Bases:
BinaryToIntDecoderThis performs Gray encoding when converting from binary strings.
See also: https://en.wikipedia.org/wiki/Gray_code#Converting_to_and_from_Gray_code
For example, a grey encoded Boolean representation of [1, 8, 4] can be decoded like this:
>>> import numpy as np >>> d = BinaryToIntGreyDecoder(4, 4, 4) >>> b = np.array([0,0,0,1, 1, 1, 0, 0, 0, 1, 1, 0]) >>> d.decode(b) array([1, 8, 4])
- decode(genome, *args, **kwargs)
Converts a Boolean genome to an integer-vector phenome by interpreting each segment of the genome as low-endian binary number.
- Parameters
genome – a list of 0s and 1s representing a Boolean genome
- Returns
a corresponding list of ints representing the integer-vector phenome
For example, a Boolean representation of [1, 12, 5] can be decoded like this:
>>> import numpy as np >>> d = BinaryToIntDecoder(4, 4, 4) >>> b = np.array([0,0,0,1, 1, 1, 0, 0, 0, 1, 1, 0]) >>> d.decode(b) array([ 1, 12, 6])
- class leap_ec.binary_rep.decoders.BinaryToRealDecoder(*segments)
Bases:
BinaryToRealDecoderCommon
- class leap_ec.binary_rep.decoders.BinaryToRealDecoderCommon(*segments)
Bases:
DecoderCommon implementation for binary to real decoders.
The base classes BinaryToRealDecoder and BinaryToRealGreyDecoder differ by just the underlying binary to integer decoder. Most all the rest of the binary integer to real-value decoding is the same, hence this class.
- decode(genome, *args, **kwargs)
Convert a list of binary values into a real-valued vector.
- class leap_ec.binary_rep.decoders.BinaryToRealGreyDecoder(*segments)
Bases:
BinaryToRealDecoderCommon
Used to initialize binary sequences
- leap_ec.binary_rep.initializers.create_binary_sequence(length)
A closure for initializing a binary sequences for binary genomes.
- Parameters
length – how many genes?
- Returns
a function that, when called, generates a binary vector of given length
E.g., can be used for Individual.create_population
>>> from leap_ec.decoder import IdentityDecoder >>> from . problems import MaxOnes >>> population = Individual.create_population(10, create_binary_sequence(length=10), ... decoder=IdentityDecoder(), ... problem=MaxOnes())
Binary representation specific pipeline operators.
- leap_ec.binary_rep.ops.genome_mutate_bitflip(genome: ndarray = '__no__default__', expected_num_mutations: float = None, probability: float = None) ndarray
Perform bitflip mutation on a particular genome.
This function can be used by more complex operators to mutate a full population (as in mutate_bitflip), to work with genome segments (as in leap_ec.segmented.ops.apply_mutation), etc. This way we don’t have to copy-and-paste the same code for related operators.
- Parameters
genome – of binary digits that we will be mutating
expected_num_mutations – on average how many mutations are we expecting?
- Returns
mutated genome
- leap_ec.binary_rep.ops.mutate_bitflip(next_individual: Iterator = '__no__default__', expected_num_mutations: float = None, probability: float = None) Iterator
Perform bit-flip mutation on each individual in an iterator (population).
This assumes that the genomes have a binary representation.
>>> from leap_ec.individual import Individual >>> from leap_ec.binary_rep.ops import mutate_bitflip >>> import numpy as np
>>> original = Individual(np.array([1, 1])) >>> op = mutate_bitflip(expected_num_mutations=1) >>> pop = iter([original]) >>> mutated = next(op(pop))
- Parameters
next_individual – to be mutated
expected_num_mutations – on average how many mutations done (specificy either this or probability, but not both)
probability – the probability of mutating any given gene (specificy either this or expected_num_mutations, but not both)
- Returns
mutated individual
- leap_ec.binary_rep.ops.random() x in the interval [0, 1).
Real-valued representations
Another common representation is a vector of real-values.
Initializers for real values.
- leap_ec.real_rep.initializers.create_real_vector(bounds)
A closure for initializing lists of real numbers for real-valued genomes, sampled from a uniform distribution.
Having a closure allows us to just call the returned function N times in Individual.create_population().
TODO Allow either a single tuple or a test_sequence of tuples for bounds. —Siggy
- Parameters
bounds – a list of (min, max) values bounding the uniform sampline of each element
- Returns
A function that, when called, generates a random genome.
E.g., can be used for Individual.create_population()
>>> from leap_ec.decoder import IdentityDecoder >>> from . problems import SpheroidProblem >>> bounds = [(0, 1), (0, 1), (-1, 100)] >>> population = Individual.create_population(10, create_real_vector(bounds), ... decoder=IdentityDecoder(), ... problem=SpheroidProblem())
Pipeline operators for real-valued representations
- leap_ec.real_rep.ops.apply_hard_bounds(genome, hard_bounds)
A helper that ensures that every gene is contained within the given bounds.
- Parameters
genome – list of values to apply bounds to.
hard_bounds – if a (low, high) tuple, the same bounds will be used for every gene. If a list of tuples is given, then the ith bounds will be applied to the ith gene.
Both sides of the range are inclusive:
>>> genome = np.array([0, 10, 20, 30, 40, 50]) >>> apply_hard_bounds(genome, hard_bounds=(20, 40)) array([20, 20, 20, 30, 40, 40])
Different bounds can be used for each locus by passing in a list of tuples:
>>> bounds= [ (0, 1), (0, 1), (50, 100), (50, 100), (0, 100), (0, 10) ] >>> apply_hard_bounds(genome, hard_bounds=bounds) array([ 0, 1, 50, 50, 40, 10])
- leap_ec.real_rep.ops.genome_mutate_gaussian(genome='__no__default__', std: float = '__no__default__', expected_num_mutations='__no__default__', bounds: Tuple[float, float] = (-inf, inf), transform_slope: float = 1.0, transform_intercept: float = 0.0)
Perform Gaussian mutation directly on real-valued genes (rather than on an Individual).
This used to be inside mutate_gaussian, but was moved outside it so that leap_ec.segmented.ops.apply_mutation could directly use this function, thus saving us from doing a copy-n-paste of the same code to the segmented sub-package.
- Parameters
genome – of real-valued numbers that will potentially be mutated
std – the mutation width—either a single float that will be used for all genes, or a list of floats specifying the mutation width for each gene individually.
expected_num_mutations – on average how many mutations are expected
- Returns
mutated genome
- leap_ec.real_rep.ops.mutate_gaussian(next_individual: Iterator = '__no__default__', std='__no__default__', expected_num_mutations: Union[int, str] = None, bounds=(-inf, inf), transform_slope: float = 1.0, transform_intercept: float = 0.0) Iterator
Mutate and return an Individual with a real-valued representation.
This operators on an iterator of Individuals:
>>> from leap_ec.individual import Individual >>> from leap_ec.real_rep.ops import mutate_gaussian >>> import numpy as np >>> pop = iter([Individual(np.array([1.0, 0.0]))])
Mutation can either use the same parameters for all genes:
>>> op = mutate_gaussian(std=1.0, expected_num_mutations='isotropic', bounds=(-5, 5)) >>> mutated = next(op(pop))
Or we can specify the std and bounds independently for each gene:
>>> pop = iter([Individual(np.array([1.0, 0.0]))]) >>> op = mutate_gaussian(std=[0.5, 1.0], ... expected_num_mutations='isotropic', ... bounds=[(-1, 1), (-10, 10)] ... ) >>> mutated = next(op(pop))
- Parameters
next_individual – to be mutated
std – standard deviation to be equally applied to all individuals; this can be a scalar value or a “shadow vector” of standard deviations
expected_num_mutations – if an int, the expected number of mutations per individual, on average. If ‘isotropic’, all genes will be mutated.
bounds – to clip for mutations; defaults to (- ∞, ∞)
- Returns
a generator of mutated individuals.
Integer representations
A vector of all integer values is also a common representation.
Initializers for integer-valued genomes.
- leap_ec.int_rep.initializers.create_int_vector(bounds)
A closure for initializing lists of integers for int-vector genomes, sampled from a uniform distribution.
Having a closure allows us to just call the returned function N times in Individual.create_population().
TODO Allow either a single tuple or a sequence of tuples for bounds. —Siggy
- Parameters
bounds – a list of (min, max) values bounding the uniform sampline of each element
- Returns
A function that, when called, generates a random genome.
>>> from leap_ec.decoder import IdentityDecoder >>> from leap_ec.real_rep.problems import SpheroidProblem >>> bounds = [(0, 1), (-5, 5), (-1, 100)] >>> population = Individual.create_population(10, create_int_vector(bounds), ... decoder=IdentityDecoder(), ... problem=SpheroidProblem())
Evolutionary operators for maniuplating integer-vector genomes.
- leap_ec.int_rep.ops.genome_mutate_binomial(std='__no__default__', bounds: list = '__no__default__', expected_num_mutations: float = None, probability: float = None, n: int = 10000)
Perform additive binomial mutation of a particular genome.
>>> import numpy as np >>> genome = np.array([42, 12]) >>> bounds = [(0,50), (-10,20)] >>> genome_op = genome_mutate_binomial(std=0.5, bounds=bounds, ... expected_num_mutations=1) >>> new_genome = genome_op(genome)
- leap_ec.int_rep.ops.individual_mutate_randint(genome='__no__default__', bounds: list = '__no__default__', expected_num_mutations=None, probability=None)
Perform random-integer mutation on a particular genome.
>>> import numpy as np >>> genome = np.array([42, 12]) >>> bounds = [(0,50), (-10,20)] >>> new_genome = individual_mutate_randint(genome, bounds, expected_num_mutations=1)
- Parameters
genome – test_sequence of integers to be mutated
bounds – test_sequence of bounds tuples; e.g., [(1,2),(3,4)]
expected_num_mutations – on average how many mutations done (specificy either this or probability, but not both)
probability – the probability of mutating any given gene (specificy either this or expected_num_mutations, but not both)
- leap_ec.int_rep.ops.mutate_binomial(next_individual: Iterator = '__no__default__', std: float = '__no__default__', bounds: list = '__no__default__', expected_num_mutations: float = None, probability: float = None, n: int = 10000) Iterator
Mutate genes by adding an integer offset sampled from a binomial distribution centered on the current gene value.
This is very similar to applying additive Gaussian mutation and then rounding to the nearest integer, but does so in a way that is more natural for integer-valued genes.
- Parameters
std (float) – standard deviation of the binomial distribution
bounds – list of pairs of hard bounds to clip each gene by (to prevent mutation from carrying a gene value outside an allowed range)
expected_num_mutations – on average how many mutations done (specificy either this or probability, but not both)
probability – the probability of mutating any given gene (specificy either this or expected_num_mutations, but not both)
n (int) – the number of “coin flips” to use in the binomial process (defaults to 10000)
Usage example:
>>> from leap_ec.individual import Individual >>> from leap_ec.int_rep.ops import mutate_binomial >>> import numpy as np >>> population = iter([Individual(np.array([1, 1]))]) >>> operator = mutate_binomial(std=2.5, ... bounds=[(0, 10), (0, 10)], ... expected_num_mutations=1) >>> mutated = next(operator(population))
The std parameter can also be given as a list with a value to use for each gene locus:
>>> population = iter([Individual(np.array([1, 1]))]) >>> operator = mutate_binomial(std=[2.5, 3.0], ... bounds=[(0, 10), (0, 10)], ... expected_num_mutations=1) >>> mutated = next(operator(population))
Note
The binomial distribution is defined by two parameters, n and p. Here we simplify the interface by asking instead for an std parameter, and fixing a high value of n by default. The value of p needed to obtain the given std is computed for you internally.
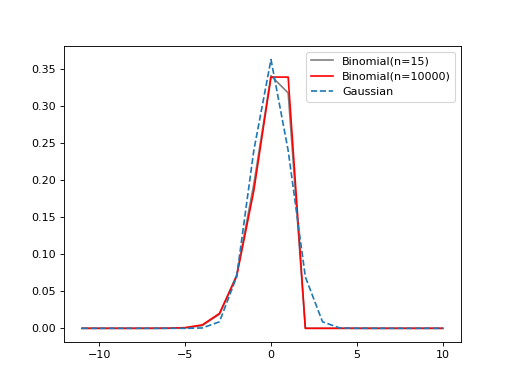
As the plots below illustrate, the binomial distribution is approximated by a Gaussian. For high n and large standard deviations, the two are effectively equivalent. But when the standard deviation (and thus binomial p parameter) is relatively small, the approximation becomes less accurate, and the binomial differs somewhat from a Gaussian.
(Source code, png, hires.png, pdf)

{kind=link}
{kind=link}
- leap_ec.int_rep.ops.mutate_randint(next_individual: Iterator = '__no__default__', bounds='__no__default__', expected_num_mutations=None, probability=None) Iterator
Perform randint mutation on each individual in an iterator (population).
This operator replaces randomly selected genes with an integer samples from a uniform distribution.
- Parameters
bounds – test_sequence of bounds tuples; e.g., [(1,2),(3,4)]
expected_num_mutations – on average how many mutations done (specificy either this or probability, but not both)
probability – the probability of mutating any given gene (specificy either this or expected_num_mutations, but not both)
>>> from leap_ec.individual import Individual >>> from leap_ec.int_rep.ops import mutate_randint >>> import numpy as np
>>> population = iter([Individual(np.array([1, 1]))]) >>> operator = mutate_randint(expected_num_mutations=1, bounds=[(0, 10), (0, 10)]) >>> mutated = next(operator(population))
Segmented representations
Segmented representations are a wrapper around another, arbitrary representation, such as a binary, real-valued, or integer representation. Segmented representations allow for sequences of value “chunks”. For example, a Pitt Approach could be implemented using this representation where each segment represents a single rule. Another example would be each segment represents associated hyper-parameters for a convolutional neural network layer.
Used to decode segments
- class leap_ec.segmented_rep.decoders.SegmentedDecoder(segment_decoder)
Bases:
DecoderFor decoding LEAP segmented representations
>>> from leap_ec.binary_rep.decoders import BinaryToIntDecoder
This example presumes that each segment has five bits, the first to map to an integer and the remaining three to a different integer.
>>> import numpy as np >>> decoder = SegmentedDecoder(BinaryToIntDecoder(2,3)) >>> genome = np.array([[1, 0, 1, 0, 1], ... [0, 0, 1, 1, 1], ... [1, 0, 0, 0, 1]]) >>> vals = decoder.decode(genome) >>> assert np.all(vals == np.array([[2, 5], [0, 7], [2, 1]]))
- decode(genome, *args, **kwargs)
For decoding genome which is a list of lists, or a segmented representation.
- Parameters
genome (will be a list of segments (or lists)) – for a given individual
args (list) – optional args
kwargs (dict) – optional keyword args
- Returns
a list of list of values decoded from genome
- Return type
list
Used to initialize segments
- leap_ec.segmented_rep.initializers.create_segmented_sequence(length, seq_initializer)
Create a segmented test_sequence
A segment is a list of lists. seq_initializer is used to create length individual segments, which allows for the using any of the pre-supplied initializers for a regular genomic test_sequence, or for making your own.
length denotes how many segments to generate. If it’s an integer, then we will create length segments. However, if it’s a function that draws from a random distribution that returns an int, we will, instead, use that to calculate the number of segments to generate.
>>> from leap_ec.binary_rep.initializers import create_binary_sequence >>> segmented_initializer = create_segmented_sequence(3, create_binary_sequence(3)) >>> segments = segmented_initializer() >>> assert len(segments) == 3
- Parameters
length (int or Callable) – How many segments?
seq_initializer (Callable) – initializer for creating individual sequences
- Returns
function that returns a list of segmented
- Return type
Callable
Segmented representation specific pipeline operators.
- leap_ec.segmented_rep.ops.add_segment(next_individual: Iterator = '__no__default__', seq_initializer: Callable = '__no__default__', probability: float = '__no__default__', append: bool = False) Iterator
Possibly add a segment to the given individual
New segments can be always appended, or randomly inserted within the individual’s genome.
TODO add a parameter for accepting a function that will yield a distribution for the number of segments to be randomly inserted.
>>> from leap_ec.individual import Individual >>> from leap_ec.binary_rep.initializers import create_binary_sequence >>> import numpy as np >>> original = Individual([np.array([0, 0]), np.array([1, 1])]) >>> mutated = next(add_segment(iter([original]), ... seq_initializer=create_binary_sequence(2), ... probability=1.0))
- Parameters
next_individual – to possibly add a segment
seq_initializer – callable for initializing any new segments
probability – likelihood of adding a segment
append – if True, always append any new segments
- Returns
yielded individual with a possible new segment
- leap_ec.segmented_rep.ops.apply_mutation(next_individual: Iterator = '__no__default__', mutator: Callable[[list, float], list] = '__no__default__') Iterator
This expects next_individual to have a segmented representation; i.e., a test_sequence of sequences. mutator will be applied separately to each sub-test_sequence.
>>> from leap_ec.binary_rep.ops import genome_mutate_bitflip >>> mutation_op = apply_mutation( ... mutator=genome_mutate_bitflip( ... expected_num_mutations=0.5 ... )) >>> import numpy as np
>>> from leap_ec.individual import Individual >>> original = Individual(np.array([[0, 0], [1, 1]])) >>> mutated = next(mutation_op(iter([original])))
- Parameters
next_individual – to possibly mutate
mutator – function to be applied to each segment in the individual’s genome; first argument is a segment, the second the expected probability of mutating each segment element.
- Returns
yielded mutated individual
- leap_ec.segmented_rep.ops.copy_segment(next_individual: Iterator = '__no__default__', probability: float = '__no__default__', append: bool = False) Iterator
with a given probability, randomly select and copy a segment
>>> from leap_ec.individual import Individual >>> import numpy as np >>> original = Individual([np.array([0, 0])]) >>> mutated = next(copy_segment(iter([original]), probability=1.0)) >>> assert np.all(mutated.genome[0] == [0, 0]) and np.all(mutated.genome[1] == [0, 0])
- param next_individual
to have a segment possibly removed
- param probability
likelihood of doing this
- param append
if True, always append any new segments
- returns
the next individual
- leap_ec.segmented_rep.ops.remove_segment(next_individual: Iterator = '__no__default__', probability: float = '__no__default__') Iterator
for some chance, remove a segment
Nothing happens if the individual has a single segment; i.e., there is no chance for an empty individual to be returned.
>>> from leap_ec.individual import Individual >>> import numpy as np >>> original = Individual([np.array([0, 0]), np.array([1, 1])]) >>> mutated = next(remove_segment(iter([original]), probability=1.0)) >>> assert np.all(mutated.genome[0] == [0, 0]) or np.all(mutated.genome[0] == [1, 1])
- param next_individual
to have a segment possibly removed
- param probability
likelihood of removing a segment
- returns
the next individual
- leap_ec.segmented_rep.ops.segmented_mutate(next_individual: Iterator = '__no__default__', mutator_functions: list = '__no__default__')
A mutation operator that applies a different mutation operator to each segment of a segmented genome.
Mixed representations
There is currently no explicit support for mixed representations, but there are plans to implement such at some point. There are a few strategies for implementing mixed values:
use a binary representation with an associated Decoder that decodes values into desired target value formats, such as sequences that are a blend of integers, floating point, and categorical variables
use a floating point representation that has an associated decoder for mapping certain floating point values to integer or categorical values; an associated mutation function may be necessary to implement pertubations that make sense for individual genes
likewise use an integer representation with tailored associated decoders and mutators to decode and change values in a bespoke way
Representation convenience class
Since the notion of a representation includes how individuals are created,
how they’re decoded, and are bound to a particular class for
an individual, the class leap_ec.representation.Representation
was created to bundle those together. By default, the class for individual is
leap_ec.individual.Individual, but can of course be any of its
subclasses.
The leap_ec.representation.Representation is used in
leap_ec.algorithm.generational_ea. In the future this may become
a formal python dataclass and be more integrated into LEAP in other functions.
A Representation is a simple data structure that wraps the components needed to define, initialize, and decode individuals.
This just serves as some syntactic sugar when we are specifying algorithms—so that representation-related components are grouped together and clearly labeled Representation.
- class leap_ec.representation.Representation(initialize, decoder=IdentityDecoder(), individual_cls=<class 'leap_ec.individual.Individual'>)
Bases:
objectSyntactic sugar for some of the monolithic functions that conveniently combines a decoder, initializer, and an Individual class since those always work in tandem, but can still be loosely coupled.
- create_individual(problem)
Make a single individual.
- create_population(pop_size, problem)
make a new population
- Parameters
pop_size – how many individuals should be in the population
problem – to be solved
- Returns
a population of individual_cls individuals